Mapping high-throughput data onto networks, modelling bacteria in 3D Colonies, and developing algorithms for Molecular Dynamics

Check out our highlights from the PLOS Computational Biology July issue:
PCSF: An R-package for network-based interpretation of high-throughput data
With recent technological developments a vast amount of high-throughput data has been profiled in an attempt at understanding the mechanism of complex diseases. The current bioinformatics challenge is to interpret the data and underlying biology, where efficient algorithms for analyzing heterogeneous high-throughput data using biological networks are becoming increasingly valuable. In this paper, Ivo Kwee and colleagues propose a software package based on the Prize-collecting Steiner Forest graph optimization approach. The PCSF package performs fast and user-friendly network analysis of high-throughput data by mapping the data onto biological networks such as protein-protein interaction, gene-gene interaction or any other correlation- or coexpression-based networks. Using the interaction networks as a template, it determines high-confidence subnetworks relevant to the data, potentially leading to predictions of functional units. It also interactively visualizes the resulting subnetwork with functional enrichment analysis.
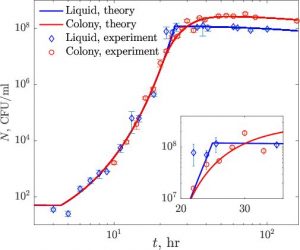
Growth of bacteria in 3D colonies
The vast majority of theoretical and experimental studies assume that bacteria exist as planktonic cells in well-mixed liquid cultures, all with equal access to nutrients, wastes, toxins, antibiotics, bacterial viruses, and each other. However, in the real world, bacteria are more often found in physically structured, spatially heterogeneous habitats as colonies and micro-colonies. While one can experimentally explore the population and evolutionary dynamics of bacteria in such physically structured habitats, there is dearth of mathematical models to generate hypotheses for and to interpret results of these experiments. As a step towards the construction of a theory of the population dynamics of bacteria in physical structured habitats, Ilya Nemenman and colleagues develop and experientially explore the simplest such model of the dynamics of bacterial growth in 3-d structured colonies.
OpenMM 7: Rapid development of high performance algorithms for molecular dynamics
OpenMM is a molecular dynamics simulation toolkit with a unique focus on extensibility. It allows users to easily add new features, including forces with novel functional forms, new integration algorithms, and new simulation protocols. Those features automatically work on all supported hardware types (including both CPUs and GPUs) and perform well on all of them. In many cases they require minimal coding, just a mathematical description of the desired function. They also require no modification to OpenMM itself and can be distributed independently of OpenMM. Peter Eastman and colleagues created this as an ideal tool for researchers developing new simulation methods, and also allows those new methods to be immediately available to the larger community.
Don’t forget to nominate your favourite 2016 PLOS Computational Biology Research Article!