Automated quaternary structure determination, computational antibody design, and cell-tracking in developing embryos

Check out our Editors-in-Chief’s selection of papers from the April issue of PLOS Computational Biology.
Automated evaluation of quaternary structures from protein crystals
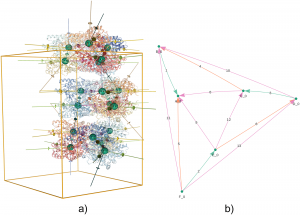
X-ray diffraction experiments are the main experimental technique used to reveal the detailed atomic three-dimensional structure of proteins. In these experiments, proteins are packed into crystals, an environment that is remote from their native solution environment. Determining which parts of the structure reflect the protein’s state in the cell rather than being artefacts of the crystal environment can be a difficult task. How the different protein subunits assemble together in solution is known as the quaternary structure. Identifying the correct quaternary structure is important to understand both protein oligomerization and protein-protein interactions in general. Here Jose M. Duarte and colleagues present a new method for automatically determining the quaternary structure of proteins given their crystal structure. They provide a theoretical basis for the properties that correct protein assemblies should possess, and provide a systematic evaluation of all possible assemblies according to these properties. The method provides guidance to the experimental structural biologist as well as to structural bioinformaticians analyzing protein structures en masse. Assemblies are provided for all proteins in the Protein Data Bank through a public website and database that is updated weekly as new structures are released.
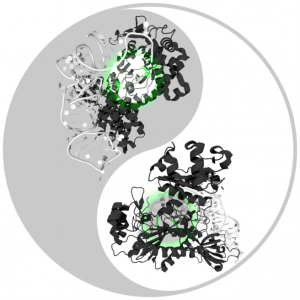
RosettaAntibodyDesign (RAbD): A general framework for computational antibody design
Antibodies are proteins produced by the immune system to attack infections and cancer, and are also used as drugs to treat cancer and autoimmune diseases. The mechanism that has evolved to produce them is able to make tens of millions of different antibodies, each with a different surface used to bind the foreign or mutated molecule. Roland L. Dunbrack Jr. and colleagues have developed a method to design antibodies computationally, based on the thousands of experimentally determined three-dimensional structures of antibodies available. The method works by treating pieces of these structures as a collection of parts that can be combined in new ways to make better antibodies. The method has been implemented in the protein modeling program Rosetta, and is called RosettaAntibodyDesign (RAbD). They tested RAbD both computationally and experimentally. The experimental test shows that they can improve existing antibodies by 10- to 50-fold, paving the way for design of entirely new antibodies in the future.
EmbryoMiner: A new framework for interactive knowledge discovery in large-scale cell tracking data of developing embryos
Kaiser et al.
Image Credit: Alexander Eisold
State-of-the-art light-sheet and confocal microscopes allow recording of entire embryos in 3D and over time (3D+t) for many hours. Fluorescently labeled structures can be segmented and tracked automatically in these terabyte-scale 3D+t images, resulting in thousands of cell migration trajectories that provide detailed insights into large-scale tissue reorganization at the cellular level. Here Benjamin Schott and colleagues present EmbryoMiner, a new interactive open-source framework suitable for in-depth analyses and comparisons of entire embryos, including an extensive set of trajectory features. Starting at the whole-embryo level, the framework can be used to iteratively focus on a region of interest within the embryo, to investigate and test specific trajectory-based hypotheses and to extract quantitative features from the isolated trajectories. Thus, the new framework provides a valuable new way to quantitatively compare corresponding anatomical regions in different embryos that were manually selected based on biological prior knowledge. As a proof of concept, they analyzed 3D+t light-sheet microscopy images of zebrafish embryos, showcasing potential user applications that can be performed using the new framework.