Exploiting protein domain co-occurrence, switching the epithelial-mesenchymal transition, optimising glioblastoma treatment, and modelling the metabolism of the malaria parasite

Check out our Editors-in-Chief’s selection of papers from the January issue of PLOS Computational Biology.
Improving pairwise comparison of protein sequences with domain co-occurrence
Deciphering the functions of the different proteins of an organism constitutes a first step toward the understanding of its biology. Because they provide strong clues regarding protein functions, domains occupy a key position among the relevant annotations that can be assigned to a protein. Protein domains are sequential motifs that are conserved along evolution and are found in different proteins and in different combinations. One common approach for identifying the domains of a protein is to run sequence-sequence comparisons with local alignment tools as BLAST. However these approaches sometimes miss several hits, especially for species that are phylogenetically distant from reference organisms. Laurent Bréhélin and colleagues here propose an approach to increase the sensitivity of pairwise sequence comparisons. This approach makes use of the fact that protein domains tend to appear with a limited number of other domains on the same protein (the domain co-occurrence property). Using the malaria pathogen Plasmodium falciparum as a case study, our approach allowed the identification of 2240 new domains for which, in most cases, no domain in the Pfam database could be ascribed.
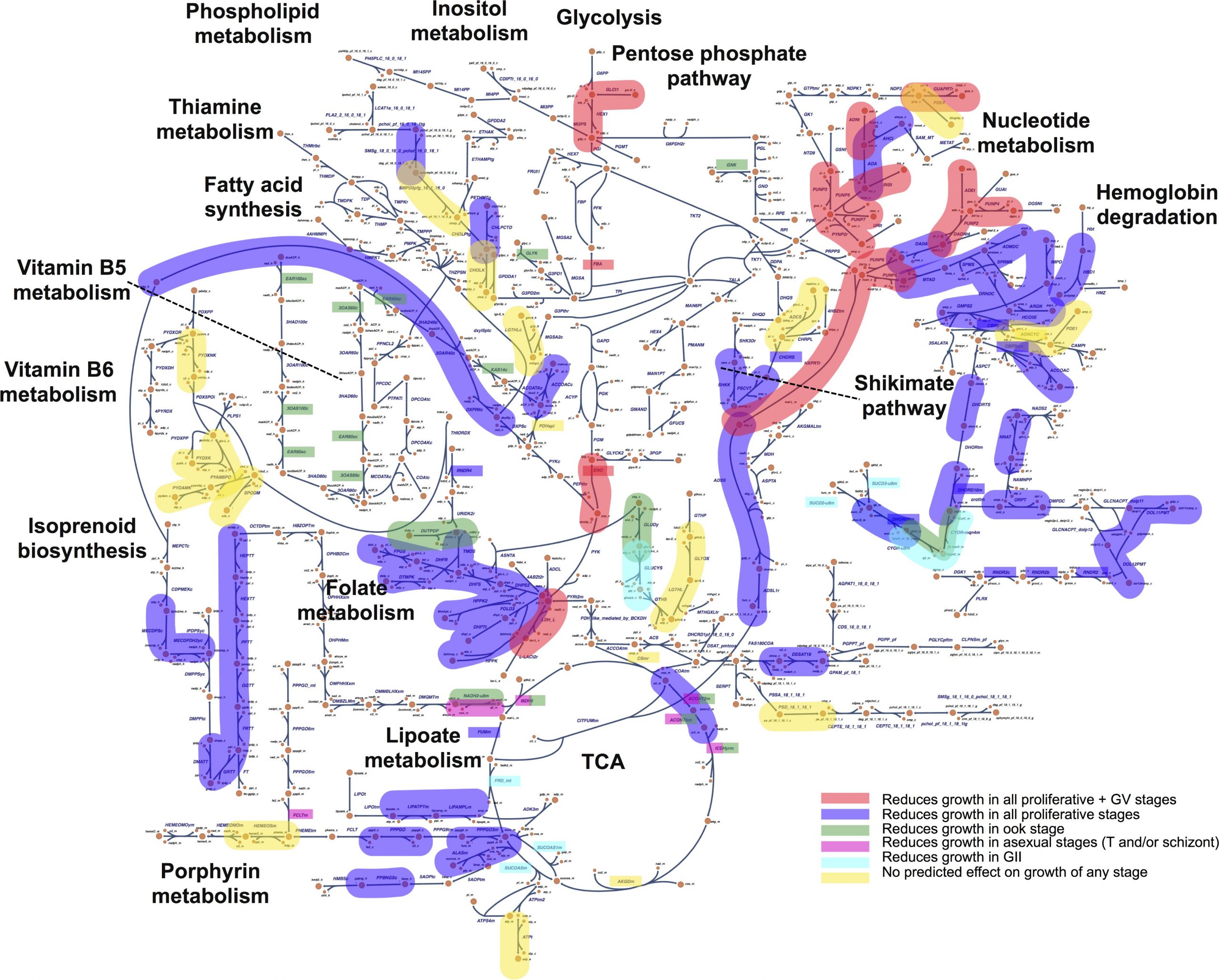
Functional interrogation of Plasmodium genus metabolism identifies species- and stage-specific differences in nutrient essentiality and drug targeting
Malaria kills nearly one-half million people a year and over 1 billion people are at risk of becoming infected by the parasite. Plasmodial infections are difficult to treat for a myriad of reasons, but the ability of the organism to remain latent in hosts and the complex life cycles have greatly contributed to the difficulty in treating malaria. Genome-scale metabolic models (GeMMs) enable hierarchical integration of disparate data types into a framework amenable to computational simulations enabling deeper mechanistic insights from high-throughput data measurements. In this study, Neema Jamshidi and colleagues used GeMMs of multiple Plasmodium species to study metabolic similarities and differences across the Plasmodium genus. In silico gene-knock out simulations across species and stages uncovered functional metabolic differences between human- and rodent-infecting species as well as across the parasite’s life-cycle stages. These findings may help identify drug regimens that are more effective in targeting human-infecting species across multiple stages of the organism.
Integration of pan-cancer transcriptomics with RPPA proteomics reveals mechanisms of epithelial-mesenchymal transition
Profiling molecular and phenotypic characteristics of large collections of cancer cell lines can be used to identify distinct and common oncogenic pathways across cancer types. So far, most large-scale data obtained from cancer cell lines have been at the genomic, transcriptomic, and phenotypic levels. Recently, high-quality data at the level of cell signaling through protein abundances and phosphorylation sites have become available. By integrating these newly generated protein data with prior transcriptomic data, and by visualizing all cancer cell lines using dimensionality reduction techniques, pan-cancer cell lines are strikingly shown to organize into a gradient of epithelial to mesenchymal types. Interestingly, many of the measured proteins and transcripts display bimodality; the expression of genes, proteins, and protein phosphorylation is either high or low, strongly suggesting that they act as molecular switches. Focusing on further characterizing molecular switches of epithelial-mesenchymal transitions, Avi Ma’ayan and colleagues identify candidate regulators and small molecules that can induce or reverse such transition, as well as potential causal relationships between proteins. Since the mesenchymal state of tumors is known to be associated with metastasis and later-stage cancer development, better understanding the regulatory mechanisms of epithelial-to-mesenchymal transition could lead to improved targeted therapeutics.
Mathematical modeling identifies optimum lapatinib dosing schedules for the treatment
of glioblastoma patients
In vivo inhibition of tumor expansion requires a sufficient amount of therapeutic agent to be present in the tumor tissue. A number of factors affect drug concentrations including the maximum tolerated dose, pharmacokinetics and pharmacodynamics profiles. Franziska Michor and colleagues present a computational modeling platform incorporating both in vitro data and published clinical trial data to investigate the efficacy of lapatinib as a function of different dosing schedules for inhibiting glioblastoma tumor cell growth. The goal of their method is to find the best dosing schedule balancing both toxicity and efficacy. The authors’ modeling approach identifies continuous dosing as the best clinically feasible strategy for slowing down tumor growth even when taking into consideration intratumor heterogeneity, drug resistance and reduced lapatinib concentrations in the tumor due to the blood brain barrier.